


Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
Initialising ...
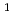 D)+N
D)+N O
O NO+NO/N+O reaction
NO+NO/N+O reaction高柳 敏幸
Chemical Physics, 308(3), p.211 - 216, 2005/01
被引用回数:6 パーセンタイル:20.35(Chemistry, Physical)O(D)+NO反応について量子-古典波束法を用いた理論計算を行った。計算は平面対称性を仮定した5次元で行い、NO分子の3振動自由度を量子波束法によって取り扱い、残りの2自由度を古典力学で取り扱った。以前われわれが開発した高精度分子軌道法の計算結果をもとにして開発した解析的なポテンシャルエネルギー曲面を用いた。この計算の目的は2つの反応生成チャンネル、NO+NO及びN+Oが衝突エネルギーやNO分子の初期振動量子状態によってどのように変化するかを理論的に調べることである。計算の結果、衝突エネルギーの増加とともに、NO+NOの生成確率が減少し、N+Oチャンネルが逆に増加することを見いだした。一方、生成分岐比はNOの初期振動量子状態によってほとんど影響を受けないことがわかった。これらの計算結果は、成層圏での熱非平衡下で起こるO(D)+NO反応のメカニズムを理解するうえで極めて重要である。
D) insertion into the C-C bond in cyclopropane and ethane黒崎 譲; 高柳 敏幸
Chemical Physics Letters, 355(5-6), p.424 - 430, 2002/04
被引用回数:3 パーセンタイル:8.63(Chemistry, Physical)シクロプロパンのC-C結合に対するO(D)挿入反応の入り口付近における5つの最低一重項ポテンシャルエネルギー面を、CASPT2/cc-pVDZレベルで計算した。その結果、5枚のポテンシャル面の内の最も下にあるものは、入り口付近で引力的であるのに対し、他の4枚は斥力的であることが予測された。比較のため、エタンについて同様の計算を行った結果、5枚のポテンシャル面は入り口付近ですべて斥力的であることが予測された。これらの計算結果は、O(D)とアルカン分子の反応についての最近の実験結果と矛盾しない。
岩本 昭; M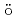 ller, P.*; Madland, D. G.*; Sierk, A.*
ller, P.*; Madland, D. G.*; Sierk, A.*
Journal of Nuclear Science and Technology, 39(4), p.332 - 336, 2002/04
被引用回数:2 パーセンタイル:16.96(Nuclear Science & Technology)核分裂における、最も生じやすい質量分割の理論計算が述べられる。模型はメッシュ点が250万以上の5次元のポテンシャルエネルギーの解析に依るものである。特別な関心は、鞍部点のエネルギーと位置を近似なしに求めることである。計算結果より、静的なポテンシャルエネルギー表面に多重の鞍部点が存在することをが示される。そのうち最も低い鞍部点とその次の鞍部点が重要であり、このうち一方が質量対称変形、残りが質量非対称変形をしていることが示される。このうちどちらが低いかは、核分裂する親核に依存して変化する。フェルミウムのアイソトープの場合には、この2者の高さは微妙に変化し、256Fmの場合には非対称変形の鞍部点が低く、一方258Fmの場合には対称変形の鞍部点が低くなる。この計算により、2重モード核分裂と呼ばれている現象が説明される。
Mller, P.*; Madland, D. G.*; Sierk, A. J.*; 岩本 昭
Nature, 409(6822), p.785 - 789, 2001/02
被引用回数:287 パーセンタイル:99.33(Multidisciplinary Sciences)核分裂のポテンシャル表面に対する、多次元の現実的な理論計算の結果が述べられる。このポテンシャル表面は、分裂核の基底状態から出発して鞍部点を超えて、最後に二つの分裂核に至る形状を記述する。今までの計算では、実験結果を再現するのに必要となる、多重の極小点や谷筋の径、尾根の存在等を記述することができる多次元のポテンシャル表面を正確に計算したものはなかった。この論文でわれわれは、5次元の形状パラメーターを動員して、260万を越すグリッド点に対する計算を行った。その結果、対称及び非対称な核分裂障壁が独立に存在すること、核分裂片の質量や相対的運動エネルギーが5次元の形状パラメーター空間での谷筋の径や尾根の存在様式と強く相関していること、を初めて明らかにした。
D)+NO reaction赤木 浩; 横山 淳; 藤村 陽*; 高柳 敏幸
Chemical Physics Letters, 324(5-6), p.423 - 429, 2000/07
被引用回数:15 パーセンタイル:43.15(Chemistry, Physical)O(D)+NO反応のポテンシャルエネルギー曲面を、高レベルの分子軌道計算により、理論的に算出した。NO分子のN原子端に、さまざまな角度方向からO(D)原子の近づくことができる。大きなポテンシャルの井戸が存在することがわかった。一方で、その接近する角度の違いで、反応経路上のエネルギー障壁の高さが変化することも判明した。これらの結果により、過去に報告されている実験結果を説明することが可能である。
NO( B
B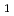 )NO(
)NO(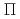 )+H reaction
)+H reaction黒崎 譲*; 高柳 敏幸
Journal of Molecular Structure; THEOCHEM, 507(1-3), p.119 - 126, 2000/07
反応HNO(2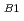 )NO()+Hの機構について非経験的分子軌道法を用いて理論的に研究した。基底状態と第一励起状態のポテンシャル面を適当な2つの内部座標の関数として、FOCI/cc-PVTZレベルで計算した。その結果、HNO(B)の分子面に垂直なCs面を保持した反応経路上に、conical intersectionが存在することが明らかとなった。同じ反応経路上には遷移状態が存在することも、既に明らかであったが、電子波動関数の対称性から、この遷移状態を経由する反応は対称禁制であることが予測されていた。しかし、今回の計算結果から、反応系はconical intersectionを回避する形で対称性をCsからCに落とすことにより禁制を解くということが明らかとなった。
)NO()+Hの機構について非経験的分子軌道法を用いて理論的に研究した。基底状態と第一励起状態のポテンシャル面を適当な2つの内部座標の関数として、FOCI/cc-PVTZレベルで計算した。その結果、HNO(B)の分子面に垂直なCs面を保持した反応経路上に、conical intersectionが存在することが明らかとなった。同じ反応経路上には遷移状態が存在することも、既に明らかであったが、電子波動関数の対称性から、この遷移状態を経由する反応は対称禁制であることが予測されていた。しかし、今回の計算結果から、反応系はconical intersectionを回避する形で対称性をCsからCに落とすことにより禁制を解くということが明らかとなった。
D)+HCN(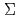 ) reaction
) reaction黒崎 譲*; 高柳 敏幸
Journal of Physical Chemistry A, 103(46), p.9323 - 9329, 1999/00
被引用回数:5 パーセンタイル:16.56(Chemistry, Physical)反応N(D)+HCN()に関して非経験的分子軌道計算を行い、有利な反応経路ならびに主生成物の理論的予測を試みた。初期過程のポテンシャル面をCASSCF(9,9)/cc-pVDZレベルで計算した結果、N(D)がHCN分子のN原子に付加する反応経路がエネルギー的に最も有利であることが予測された。中間生成物から遷移状態を経て最終生成物に至るいくつかの反応経路をPMP4(SDTQ)/cc-pVTZ//MP2/cc-pVTZレベルで計算した結果、HNCN分子が中間生成物として最も安定であり、主な最終生成物はCH+Nであることが予測された。HNCN及びCH+Nは反応物N(D)+HCN()と比較して、それぞれ117.8,53.1kcal/mol安定であることが計算された。
 ; Is a C
; Is a C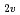 contact ion pair an isomer of Td-CCl?
contact ion pair an isomer of Td-CCl?高柳 敏幸; 黒崎 譲*; 田池川 浩人*
Radiation Physics and Chemistry, 55(4), p.367 - 371, 1999/00
被引用回数:1 パーセンタイル:13.12(Chemistry, Physical)CCl分子のポテンシャルエネルギー曲面について分子軌道計算を行った。従来の研究結果で示されたイオンペアCCl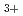 …Cl
…Cl に相当する極小値は曲面上に存在しないことがわかった。すなわちイオンペア構造は最も安定なTd構造の異性体としては存在し得ない。
に相当する極小値は曲面上に存在しないことがわかった。すなわちイオンペア構造は最も安定なTd構造の異性体としては存在し得ない。
Mller, P.*; 岩本 昭
Nuclear Physics A, 575, p.381 - 411, 1994/00
被引用回数:27 パーセンタイル:82.27(Physics, Nuclear)2つの重イオン間のポテンシャルエネルギーの計算において、双方の重イオンが変形して且つ任意の相対的配位にある場合の計算法の定式化を行う。計算は有限レンジ液滴模型に従い、変形のパラメータやエネルギーの表式を変えても容易に一般化できる形式に与えられている。この定式化に従い、重イオン反応で多次元性が問題となる幾つかの現象の解析を行った。重イオン核融合や、変形した複合核からの荷電粒子の放出において、原子核の変形がポテンシャルエネルギーにどう反映されるかが初めて定量的に与えられた。
reactions at low temperatures; Effect of rotational energy of reagent H molecule高柳 敏幸; 正木 信行
Journal of Chemical Physics, 95(6), p.4154 - 4159, 1991/09
被引用回数:13 パーセンタイル:48.18(Chemistry, Physical)H+H(j=0.1)H(j=0.1)+H反応について、系の並進エネルギーが小さい時の反応確率をJ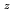 を保存する状態結合法により計算した。反応のポテンシャルエネルギー曲面としてはLSTHおよびPKを用いた。ウィグナーのしきい値則に従うようになる並進運動エネルギーは、ポテンシャルエネルギー曲面上に存在するファンデアワールスのくぼみとHの回転エネルギー状態に存在する。反応確率から低温での速度足数を計算し、固相で報告された実験値との比較を行った。反応系Hの回転エネルギーの速度定数に対する効果について議論した。
を保存する状態結合法により計算した。反応のポテンシャルエネルギー曲面としてはLSTHおよびPKを用いた。ウィグナーのしきい値則に従うようになる並進運動エネルギーは、ポテンシャルエネルギー曲面上に存在するファンデアワールスのくぼみとHの回転エネルギー状態に存在する。反応確率から低温での速度足数を計算し、固相で報告された実験値との比較を行った。反応系Hの回転エネルギーの速度定数に対する効果について議論した。
黒崎 譲; 横山 啓一
no journal, ,
本研究では、最適制御理論を用いてアルカリハライド分子の同位体選択的振動回転励起の量子制御について考察する。ここではLiCl分子の1:1同位体混合気体(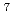 Li
Li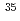 ClとLi
ClとLi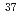 Cl)を考え、既に一方の同位体分子の回転状態がJ=20付近に励起されている状況を仮定する。この状況において、別の制御パルスにより一方の同位体分子の振動レベルを一段上げることを試みる。計算の結果、効率的に同位体選択的振動励起を実現するパルスが予測された。ここでは初期の回転励起による同位体分子間の振動遷移エネルギーの差の拡大が、より効率的な振動励起に結び付いたものと解釈できる。
Cl)を考え、既に一方の同位体分子の回転状態がJ=20付近に励起されている状況を仮定する。この状況において、別の制御パルスにより一方の同位体分子の振動レベルを一段上げることを試みる。計算の結果、効率的に同位体選択的振動励起を実現するパルスが予測された。ここでは初期の回転励起による同位体分子間の振動遷移エネルギーの差の拡大が、より効率的な振動励起に結び付いたものと解釈できる。
